宁波材料所在AI+材料计算模拟领域取得系列进展
基于量子力学的原子层级模拟计算是材料学中一种直观有效且常用的研究方法,它可以研究材料的空间原子结构、电子结构,以及由此带来的各种宏观物理、化学性质。长期以来,材料计算模拟的发展受到计算尺度的严重制约,例如描述理想周期结构、完美晶格的密度泛函理论仅可求解百原子量级的体系。然而真实的材料体系是不完美并且非常复杂的,材料中存在缺陷、晶畴界、表界面、非晶无序等结构特征,处于非平衡态的材料体系同时具有动力学演化行为,这些复杂体系的特征行为体现在更大的时间和空间尺度,因此需要大尺度的模拟计算才能描述。基于传统物理“规则驱动”的计算技术已难以从理论框架突破尺度限制。
针对这一问题,中国科学院宁波材料技术与工程研究所柔性磁电功能材料与器件团队利用并发展了AI+材料计算模拟方法。基于“数据驱动”的AI是从数据和观测值出发,寻找数据之间的特征和关系,从而发现一些定理和规律。AI与科学的结合带来了新的科研范式,给材料计算模拟带来全新的思路和视角。Deep-Potential(DP)是一种具有代表性的AI技术,它运用深度神经网络技术,采用大量小原胞(数十个原子)的密度泛函理论计算数据作为训练集,训练完成的网络可以高效准确地预测出大原胞(最高可计算百万个原子)的总能以及原子受力,从而实现大时间空间尺度(微米/纳秒)的动力学模拟。
钟志诚研究员带领研究小组近期开展了一系列DP相关的研究:1)通过研究SrTiO3的结构相变,发现了DP模型具有超高精度,与密度泛函理论计算得到的能量误差可达到meV/atom以内[Phys. Rev. B 105,064104(2022)];结合DP势函数和位错解析理论,在大尺度下准确描述Cu的位错芯结构以及位错间的长程弹性相互作用[Comput. Mater. Sci. 218,111941 (2023)]。上述两个工作证实了DP在大尺度下的高精度以及描述位错等复杂结构的有效性。2)利用DP,解释了ZrW2O8的负热膨胀现象以及压力诱导的非晶现象[Phys. Rev. B 106, 174101 (2022)],该工作表明DP势函数能够有效描述复杂动力学行为以及非晶无序结构。3)晶格量子效应对热力学等性质的求解至关重要,而却往往因为其较高的计算成本在模拟计算中往往被忽略。团队以SrTiO3的量子顺电现象为例,提出了结合DP+QTB高效地研究材料中的晶格量子效应方案[Phys. Rev. B 106, 224102 (2022)]。
以上工作为未来材料计算模拟研究提供了全新范式,为复杂材料体系的高精度大尺度模拟提供了具体思路。此外,结合AI+材料计算模拟进行大尺度及复杂效应的计算,有望解决一系列复杂材料体系中的微观机制、宏观性能等问题。例如多元体系中的高熵合金、固液界面;机制复杂的摩擦、张力、非晶、表面重构;化学反应的表面吸附、催化、燃烧等问题。
以上工作参与者包括中科院宁波材料所博士后何日、邓凤麟,博士研究生吴宏宇,合作者包括南京大学物理学院卢毅教授,西湖大学理学院刘仕教授,深势科技首席科学家张林峰博士。以上工作得到了国家重点研发计划(2021YFA0718900和2022YFA1403000)、国家自然科学基金(11974365和12204496)、中国科学院前沿科学重点研究计划(ZDBS-LY-SLH008)以及王宽诚教育基金(GJTD-2020-11)的支持。
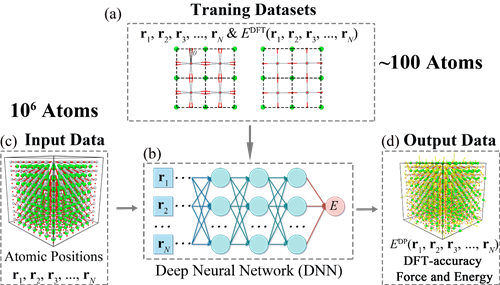
图1 (a) 通过密度泛函理论所计算的大量空间构型(约百原子级别)的能量和力;(b)DP训练所得的深度神经网络;(c)和(d)训练好的深度神经网络能应用于预测超胞(约百万原子级别)的能量和受力,其精度和密度泛函理论一致

图2 课题组近期各工作。左上:DP势函数的精度展示;右上:DP方法描述位错间对数形式的长程弹性相互作用;左下:ZrW2O8的压力诱导非晶现象;右下:DP+QTB预测的SrTiO3结构相变
(磁材实验室 吴宏宇、何日)